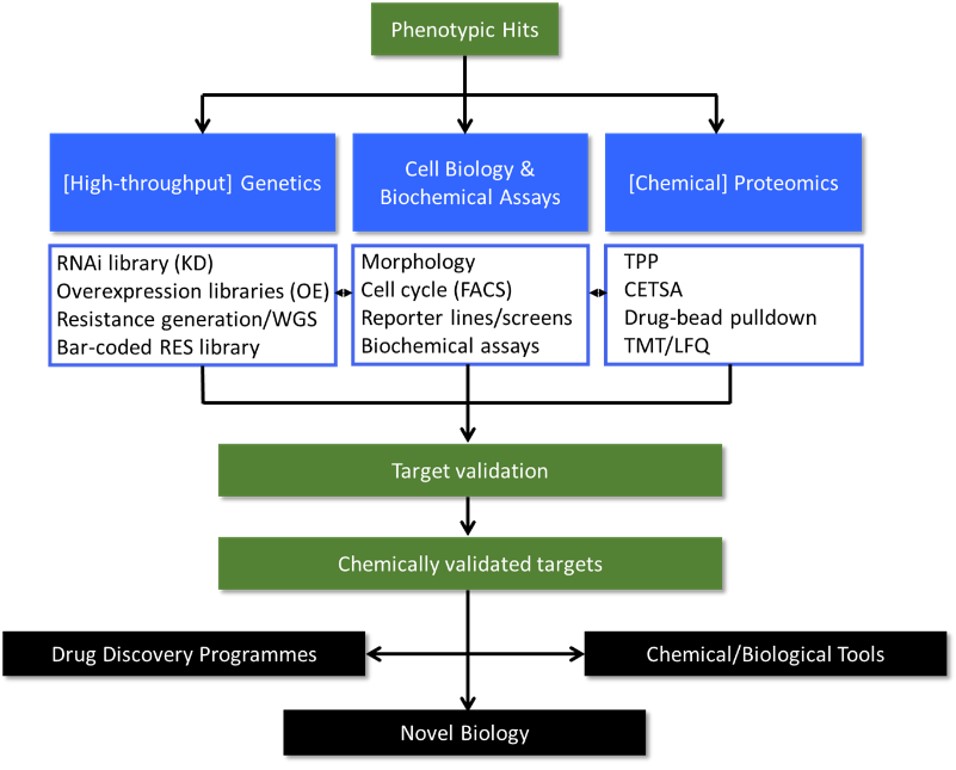
Supported by funding from the Wellcome Trust, the MoA group was established in 2015 to underpin the drug-discovery programmes within WCAIR and also works with multiple academic groups and key stakeholders in NTD drug discovery. The group is focused on determining the MoA and/or molecular targets of compounds that are phenotypically-active against parasites. In the course of our studies, we have developed an integrated drug target deconvolution strategy, employing a matrix of established and new methodologies encompassing high-throughput genetics, cell biology and chemical proteomics (Figure 1). This multi-disciplinary approach to drug target deconvolution has proven extremely effective, with the molecular targets of >65 compound series (including 5 clinical candidates) identified to date. Information emanating from these MoA studies has been invaluable in guiding anti-kinetoplastid drug discovery programmes. For example, novel drug targets have been exploited using structure-based approaches and new target-based screens, compounds that act via mechanisms unsuitable for drug development have been de-prioritised, and diverse chemical series have been identified that inhibit a particularly promiscuous molecular target. In addition, we have used our MoA toolkit for unbiased confirmation of on-target activity for compounds developed in target-focussed programmes. Owing to our success, we have recently received funding from the Bill and Melinda Gates Foundation and Medicines for Malaria Venture to support MoA studies in Plasmodium. In our new programme of research, again supported by the Wellcome Trust, we are capitalising on experience gained in recent years to substantially expand our multi-disciplinary approach to drug target deconvolution into new disease areas, specifically cryptosporidiosis and schistosomiasis.
Figure 1. Overexpression library workflow. KD, knock-down; OE, over-expression; WGS, whole genome sequencing; FACS, fluorescence activated cell sorting; CETSA, cellular thermal shift assay; TMT, tandem mass tags; LFQ, label-free quantitation.
Innovation in drug target deconvolution
To support our multi-disciplinary approach to drug target deconvolution the MoA group has established and implemented several novel methodologies.
High-throughput genetics – We have developed genome-wide overexpression libraries in all three kinetoplastid parasites (Trypanosoma brucei, Leishmania donovani and T. cruzi). These libraries are based on the principal that overexpression of a drug target may confer a selective advantage to parasites under drug selection (Figure 2). Thus, our genome-wide overexpression libraries, in combination with Next Generation Sequencing, now enable us to carry out unbiased and massive parallel screens of the parasite proteomes for the targets of drugs. We are also in the process of developing an analogous overexpression library in Plasmodium knowlesi.
Figure 2. Overexpression library workflow.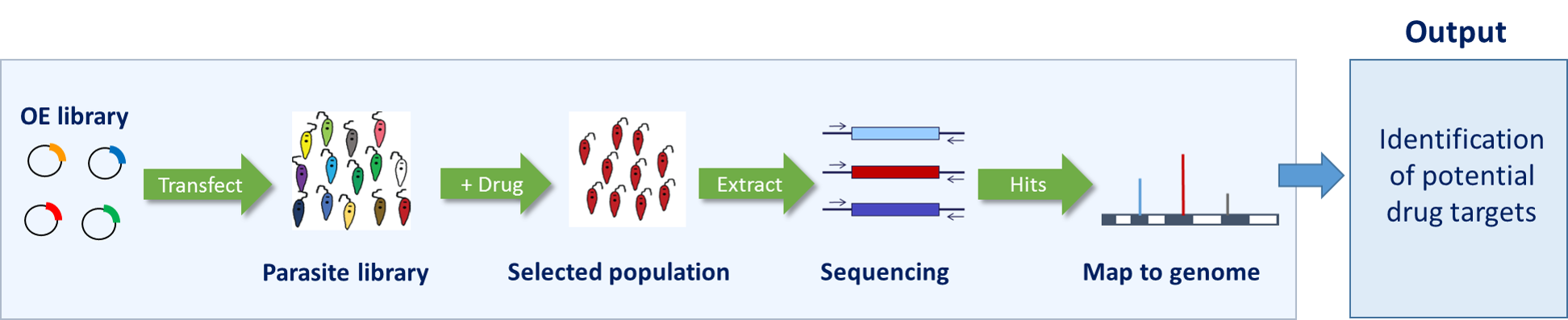
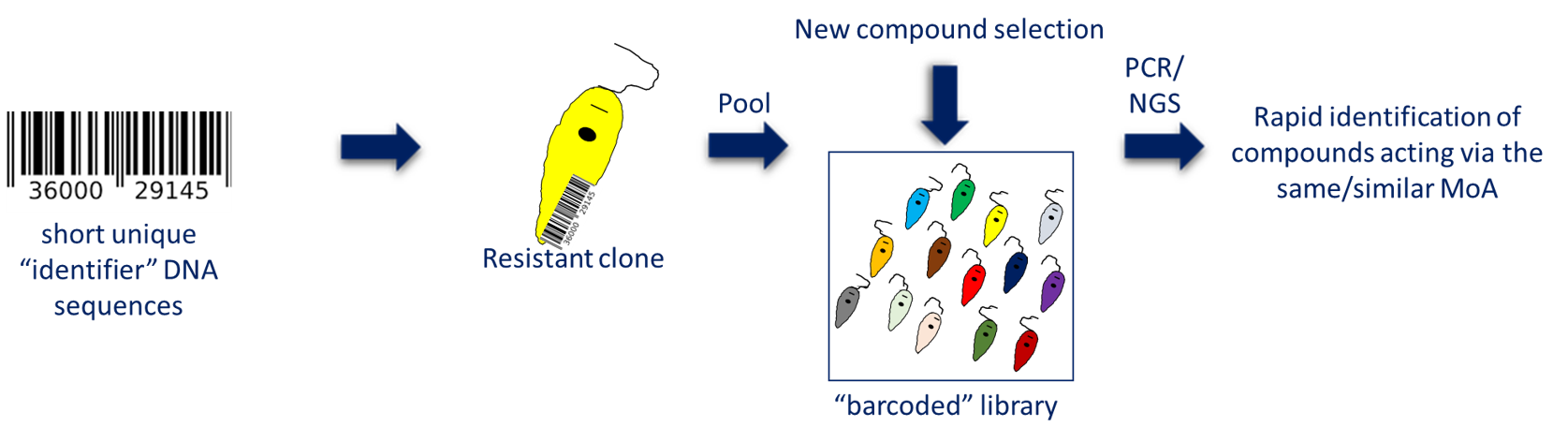
Bar-coded resistance panel – In vitro generation of compound-resistant parasites followed by WGS to identify resistance-related genomic changes is a standard MoA approach. My group have amassed a large collection of drug-resistant cell lines with defined resistance-conferring mutations (kinetoplastids). This panel of cell lines is an invaluable resource since test compounds can be screened to determine potential cross-resistance and shared MoA. To expedite screening of these cell lines, we have introduced short identifier DNA sequences or “bar-codes” into each resistant cell line allowing all lines to pooled and screened simultaneously. Cross-resistant parasites growing out following compound selection are identified by PCR amplification and sequencing of their unique barcode. This bar-coded library is now routinely and successfully used in my lab to profile new compounds that we have been asked to study.
Figure 3. Barcoded resistance panel strategy.
Cell biology and biochemistry – As a direct result of our MoA studies, a number of novel and high value drug targets have been identified that are exploitable for future drug discovery. In these cases, we have gone on to develop complementary cell-based tools and secondary biochemical assays to support subsequent drug discovery efforts against these promising targets. This is exemplified by our identification of the Leishmania proteasome as the molecular target of the clinical candidate, GSK3494245A and our subsequently development of a fluorescence-based, high-throughput cell-based assay capable of identifying inhibitors of the parasite proteasome. Similarly, in cases where we have identified undesirable targets, we have developed cell-based tools to rapidly identify and deprioritise compounds acting via mechanisms that cannot be advanced, for example chelators of divalent transition metals.
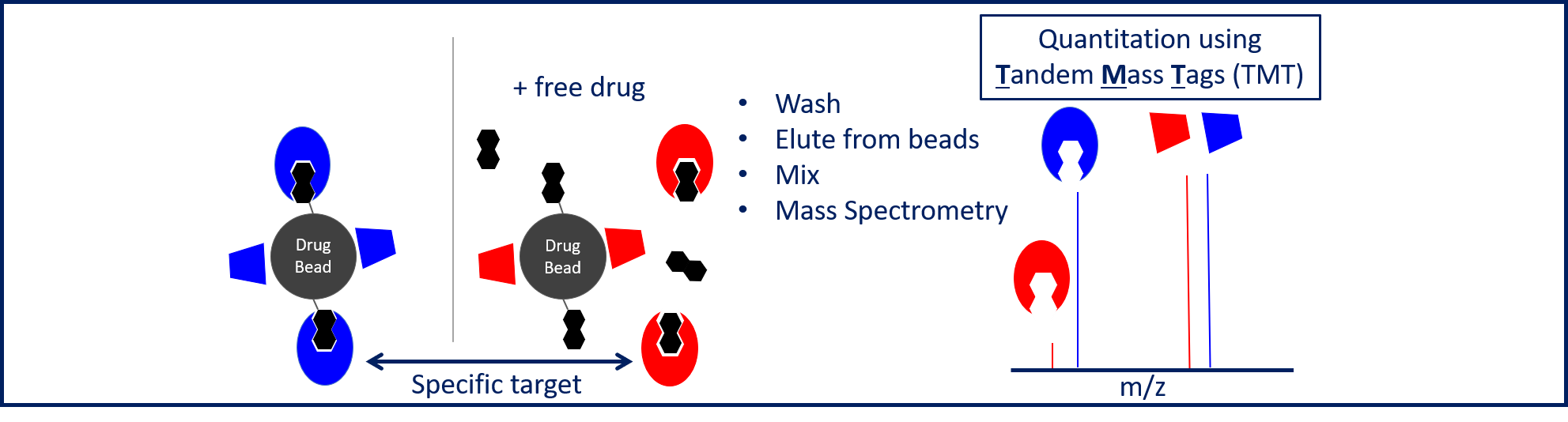
Chemical proteomics – We use compounds immobilised on magnetic beads to directly pull-down protein targets from cell extracts. To improve the discrimination of proteins that bind specifically/non-specifically to beads, we carry out pulldowns +/- competition from free compound. Targets specifically interacting with drug beads are then identified and quantified by mass spectrometry (MS). (Figure 4). Proteins specifically binding to drug beads are subsequently identified by MS and quantified by label-free quantitation.
Figure 4. Pulldown strategy.
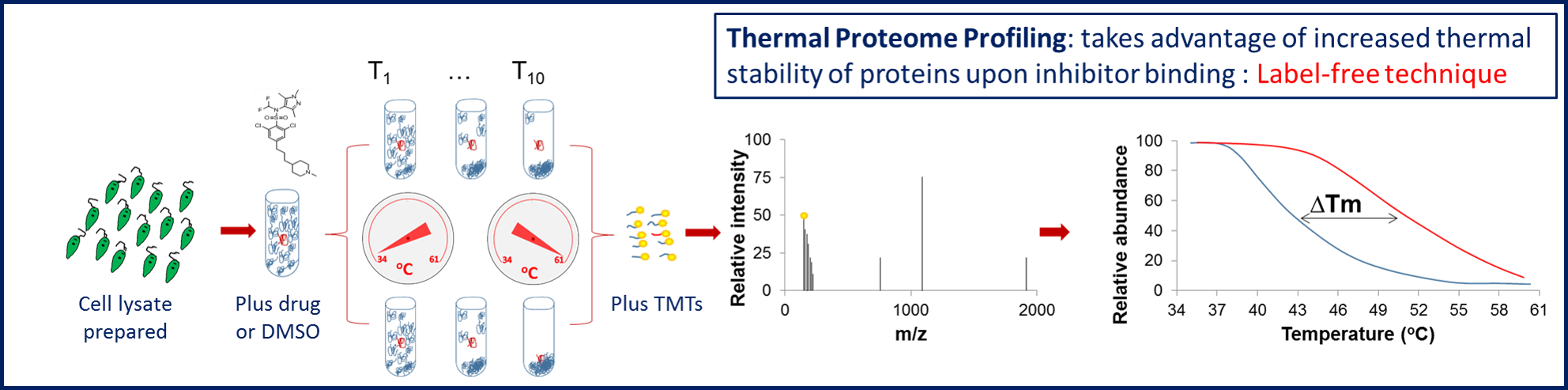
In cases where immobilisation compromises the ability of the compound to bind to its target(s), or if there is limited / no structure-activity relationship (SAR) data, we employ thermal proteome profiling (TPP) assays (Figure 5). The MoA group was the first to establish this innovative approach for use in the kinetoplastids. TPP is an unbiased, powerful label-free proteomics approach based on the principle that binding of a drug to its protein target may significantly increase the thermal stability of that protein.
Figure 5. TPP workflow.